【第一百二十二期】基层检验网形态学培训
【第一百二十二期】基层检验网形态学培训
每周一次,每次几张,只要您坚持来,抽出几分钟的时间,相信您的识图能力会不知不觉的提升哦
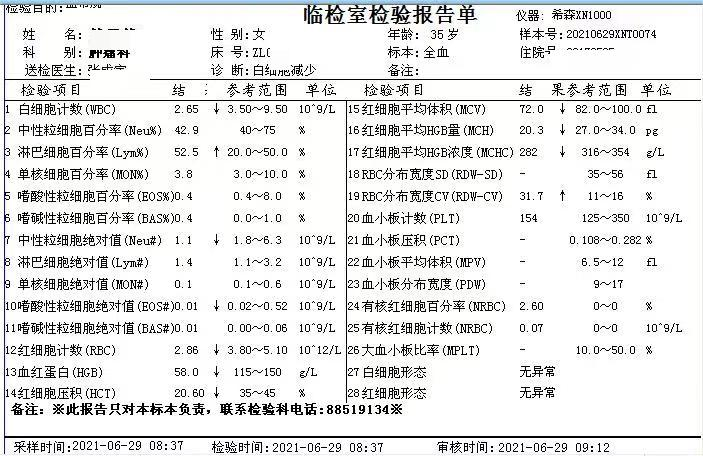
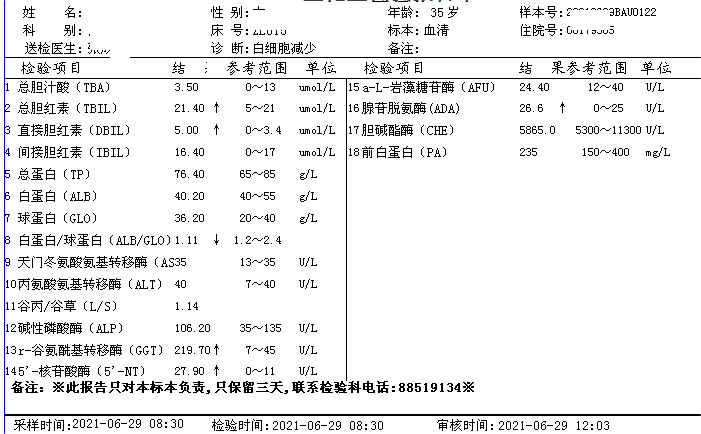
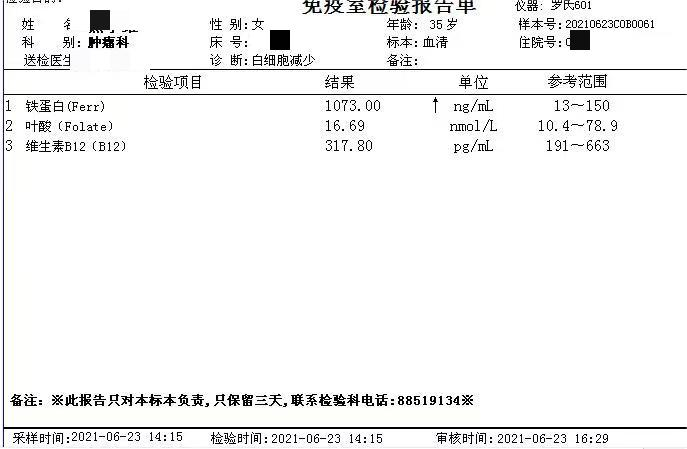
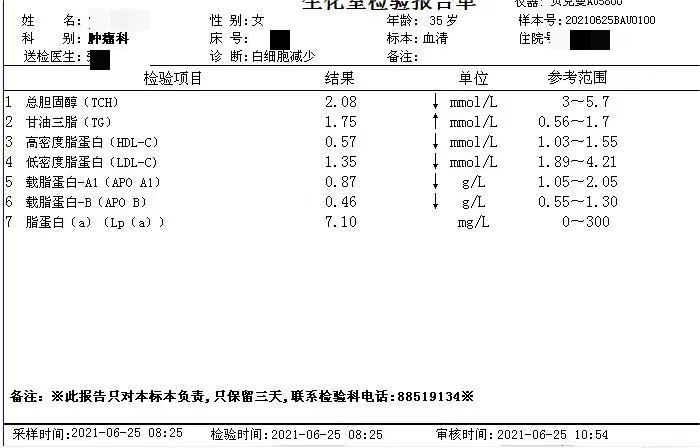
简要病史:患者青年女性,以白细胞减少,贫血原因待查入院。部分检查结果如下。查体贫血貌、肝大、脾大。

1、请写出图中细胞名称(瑞吉氏染色,1000倍油镜视野观察):
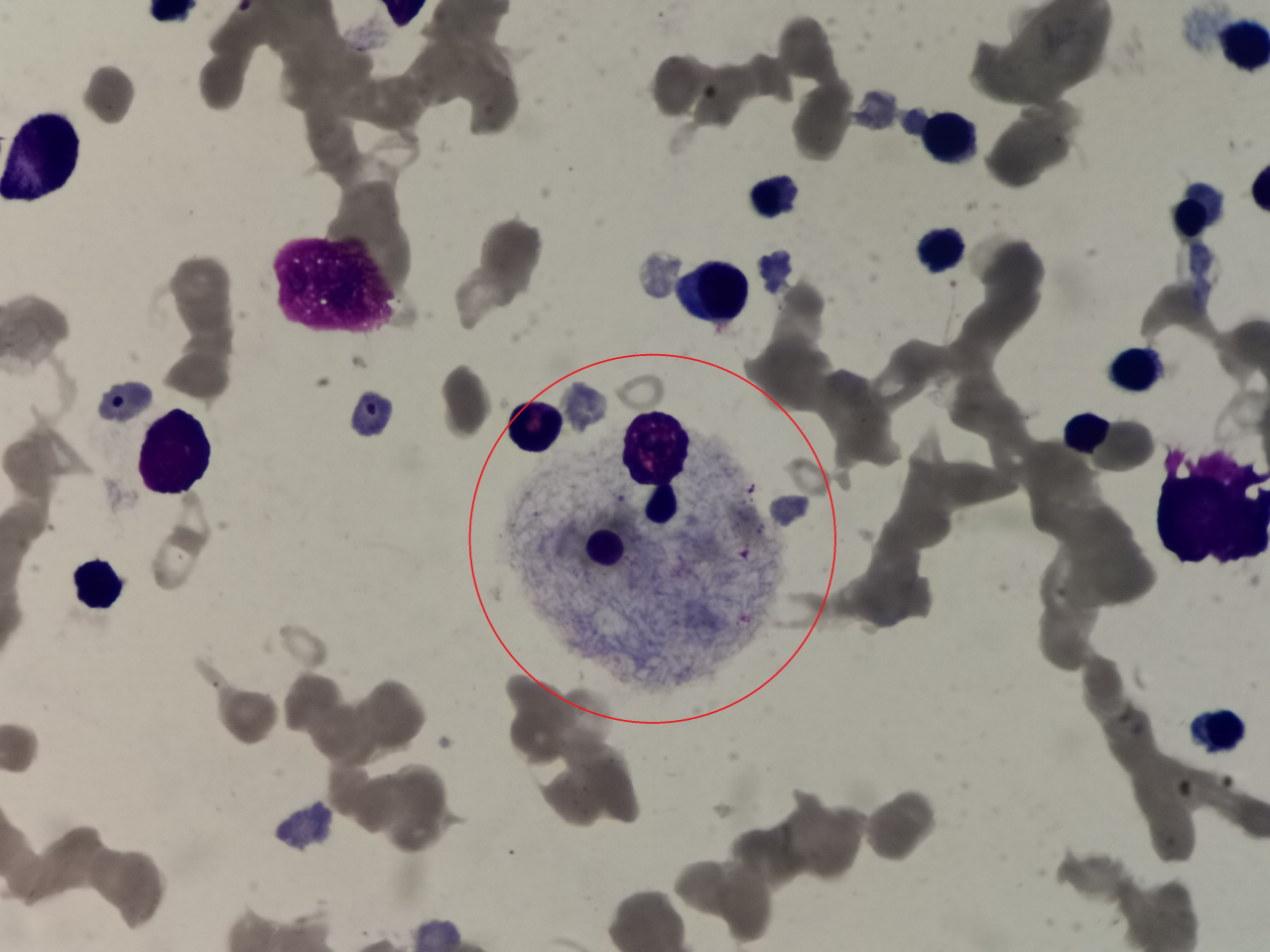
答案:尼曼匹克细胞
解析:无
2、请写出图中细胞名称(瑞吉氏染色,1000倍油镜视野观察):
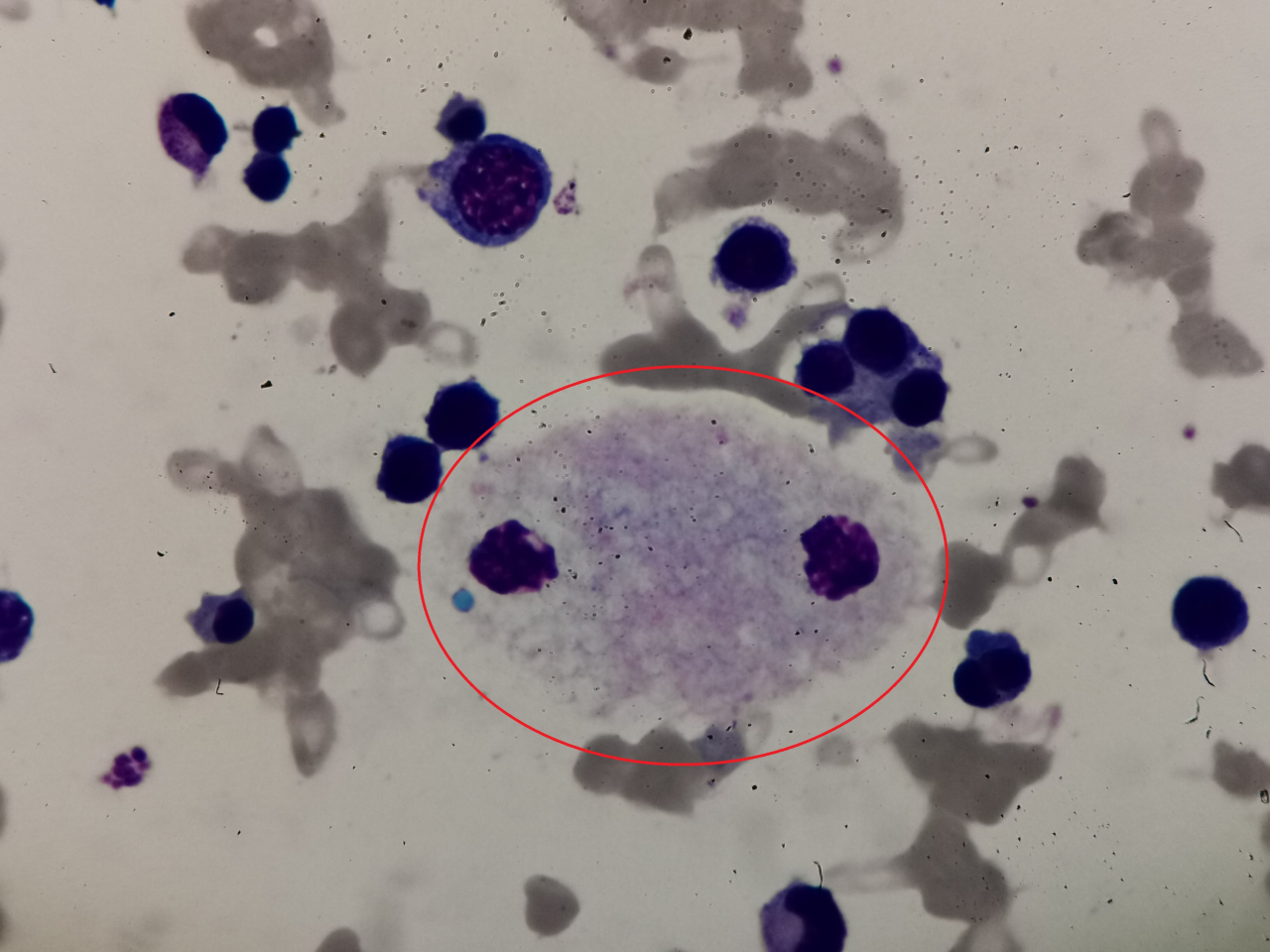
答案:尼曼匹克细胞
解析:无
3、请写出图中细胞名称(瑞吉氏染色,1000倍油镜视野观察):
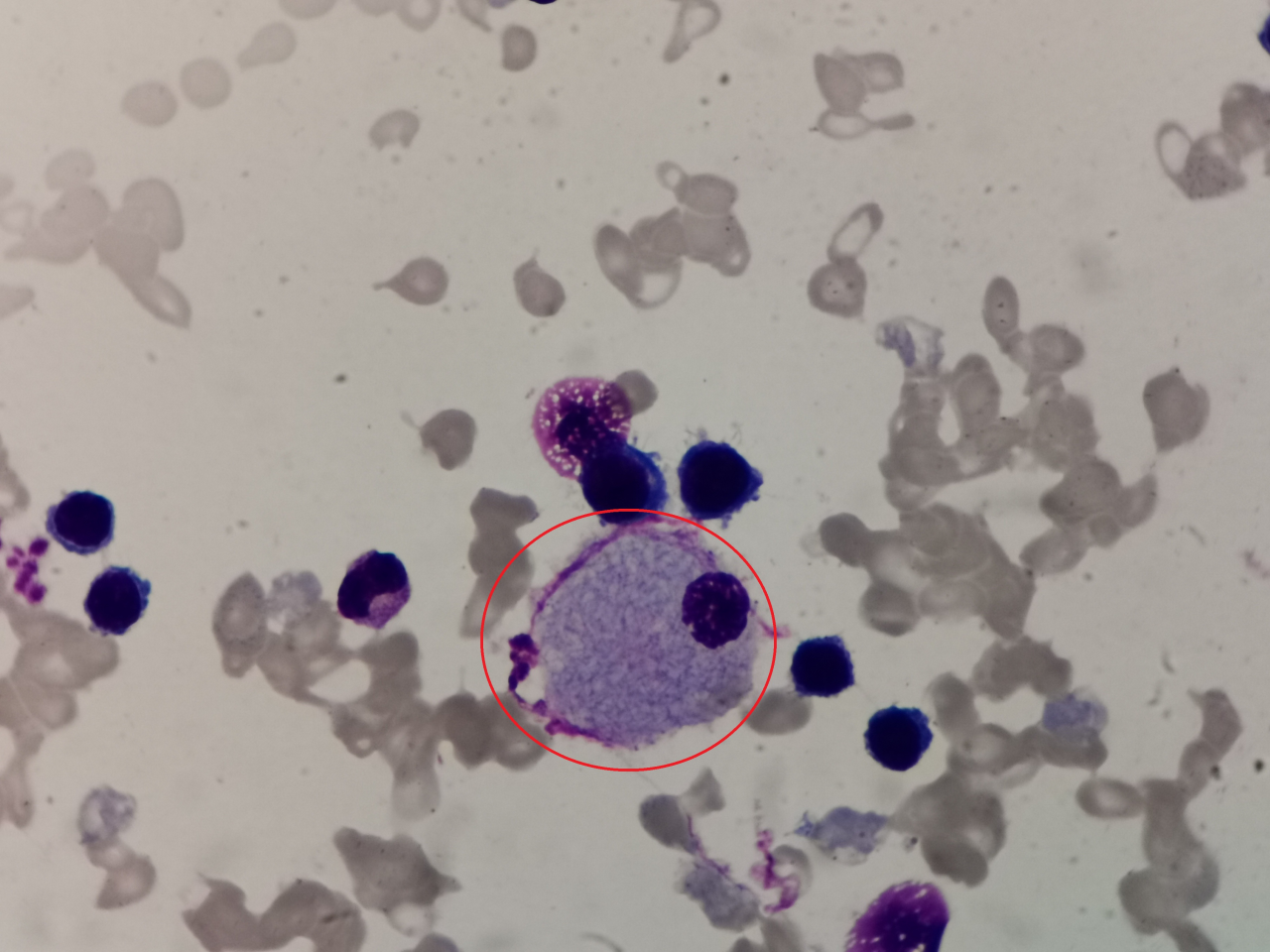
答案:尼曼匹克细胞
解析:无
4、请写出图中细胞名称(瑞吉氏染色,1000倍油镜视野观察):
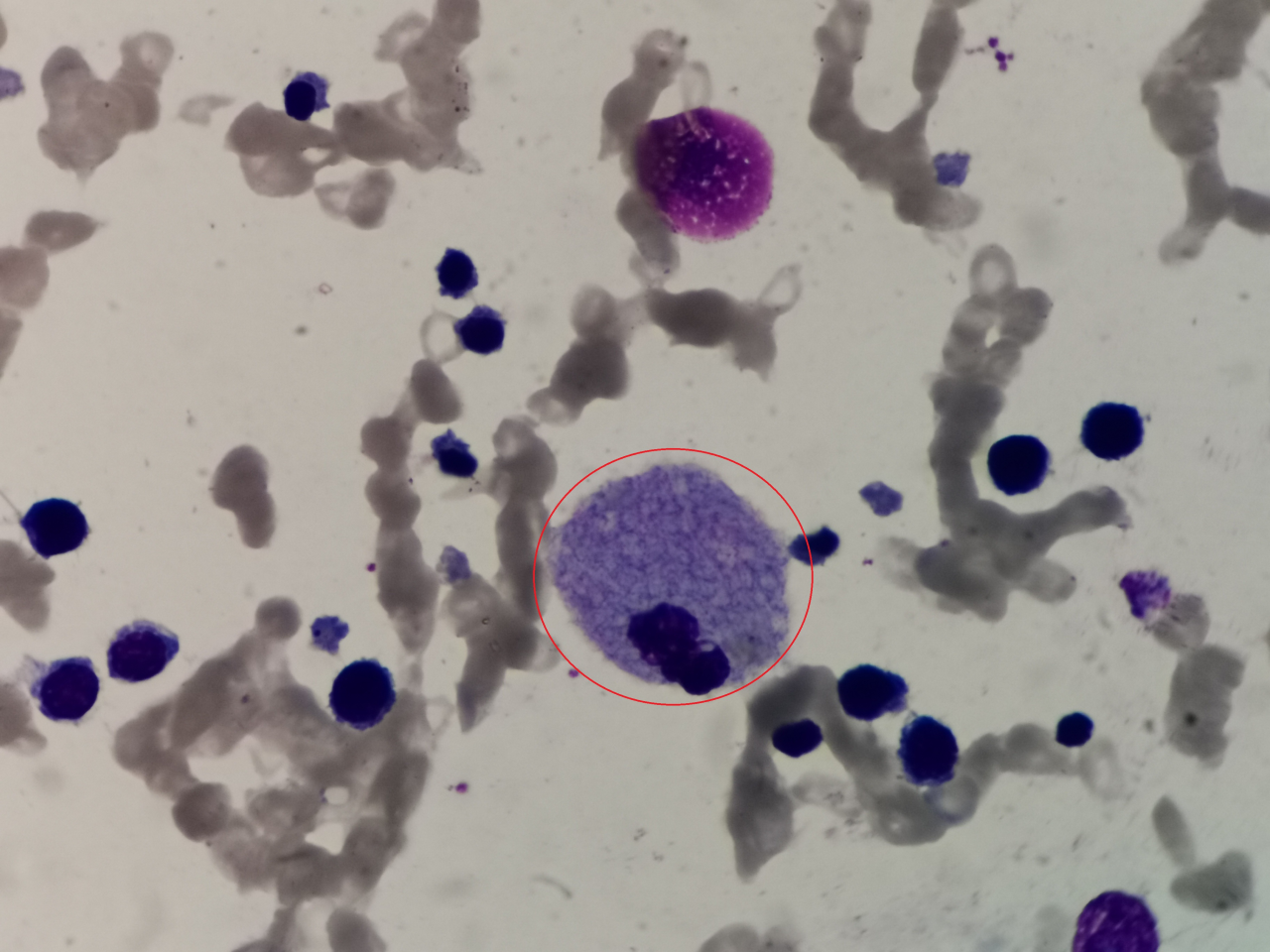
答案:尼曼匹克细胞
解析:无
5、诊断为什么病?
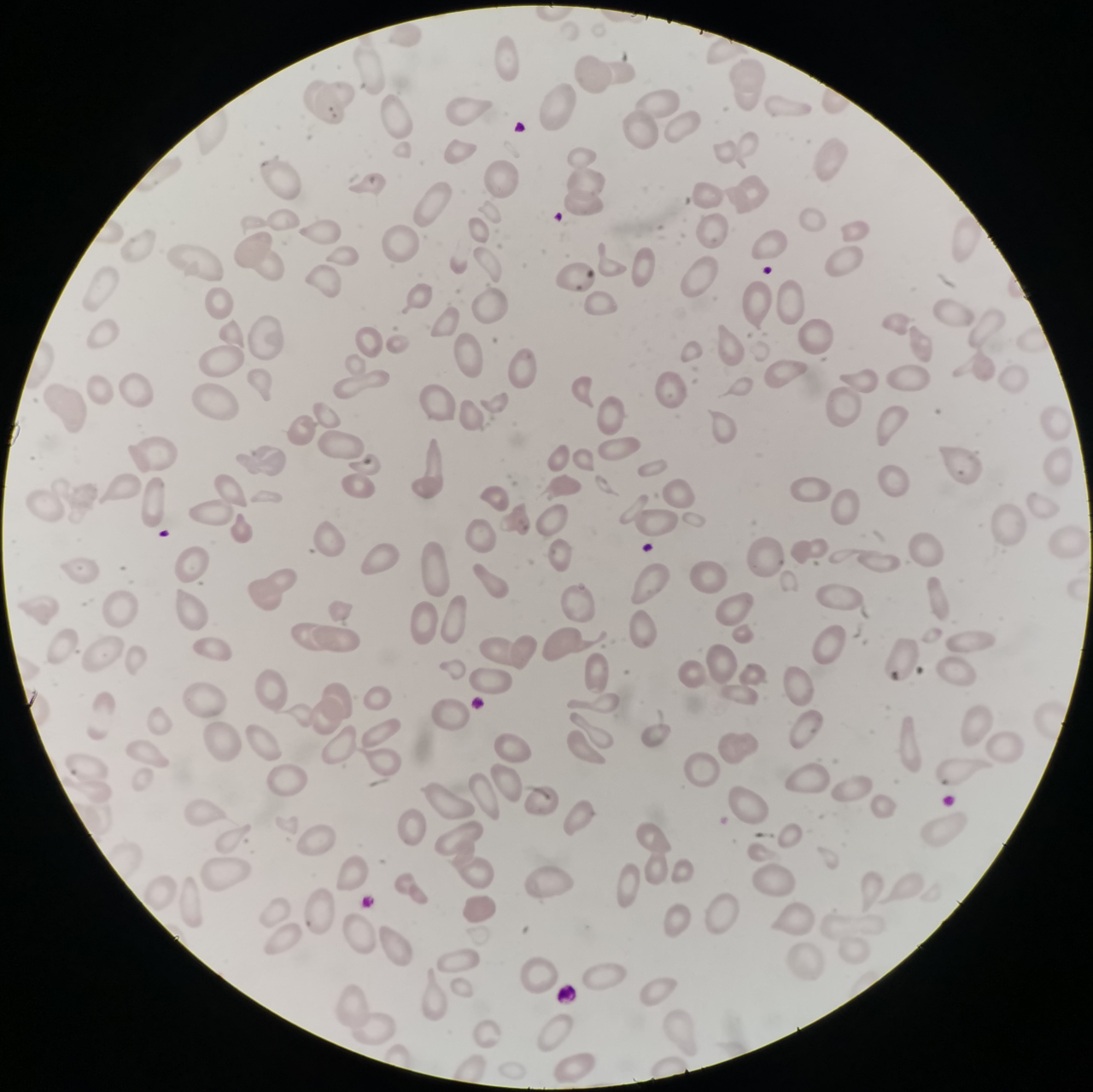
答案:尼曼-匹克病
解析:成熟红细胞体积明显大小不均,中央淡染区扩大,异形红细胞(泪滴红、裂红细胞等)易见。
尼曼匹克病又称神经鞘磷脂病,为常染色体隐性遗传病,是因为神经鞘磷脂及胆固醇沉积于身体各个器官引起纸质代谢紊乱的一种少见的家族性疾病。
【病理生理】
本病特点全身单核-巨噬细胞系统和神经系统内有大量的含有神经鞘磷脂的泡沫细胞。其发病机制是由于患者缺乏神经鞘磷脂酶导致神经鞘磷脂代谢障碍,后者在单核-巨噬细胞系统和神经组织细胞中沉积,出现肝、脾大和中枢神经系统退行性变。
【临床特征和分型】
尼曼匹克病好发于儿童,婴儿型多于2岁内死亡,成人型常为慢性型,主要症状为逐渐发生消化系统障碍,营养性贫血、肝脾,有时出现黄疸,可有智力迟钝,大多死于全身衰竭、肺部感染或脾功能亢进出血。一般分为5型,(A-E型)
1、A型(婴儿型或急性神经病变型)最常见,出生后3-6月内发病,进展快,多于3-4岁前继发感染而死亡;
2、B型(慢性非神经型)幼儿或儿童期发病,进展缓慢,肝脾大明显,智力正常;
3、C型(慢性神经型)起病较晚,垂直性核上性眼肌麻痹是特征性改变,可出现特征性眼底樱桃红斑点
4、D型(Nova Scotia型)2-4岁发病,有明显黄疸,肝脾大和神经症状,多于学龄期死亡,我国少见;
5、E型(成人非神经型)成人发病,智力正常,见不同程度肝脾大,无神经症状,可长期生存,眼底有樱桃红斑。
【细胞形态学和实验室检查】
1、血象 血红蛋白正常或轻度贫血,血小板减少,白细胞减少、正常或增高不定,单核细胞和淋巴细胞胞浆中可见特异性空泡,网织红计数正常或偏高
2、骨髓象 找到典型的尼曼匹克细胞是诊断本病的主要依据
3、细胞化学染色 苏丹三阳性、苏丹黑B及油红O染色阳性,糖原染色空泡壁阳性,铁染色阳性,酸性磷酸酶染色阴性或弱阳性,碱性磷酸酶及过氧化物酶染色阴性,非特异性酯酶染色呈阳性反应
4、组织病理学检查 肝、脾、淋巴结的活检中可见尼曼匹克细胞
5、其他检查 患者组织器官中的神经鞘磷脂含量明显增高,部分患者肝功异常,血脂代谢紊乱,通过基因分型有助于确定临床表型。
【诊断与鉴别诊断】
1、肝脾大,或婴儿期就有肝脾大,且肝大于脾,并逐渐出现神经系统表现;
2、眼底樱桃红斑,X线肺部呈粟粒样或网状浸润;
3、外周血淋巴细胞和单核细胞可见空泡,骨髓涂片查见尼曼匹克细胞是本病诊断重要依据;
4、组织活检(骨髓、肝脾、淋巴结)找到较多尼曼匹克细胞可证实;
5、神经鞘磷脂酶活性测定减低可确诊
需要与糖原贮积症、戈谢病、神经节苷脂贮积症、Wolman病、半乳糖唾液酸贮积症、原发性海蓝组织细胞增多症等鉴别诊断
免责声明:基层检验网订阅号是一个提供学习讨论、资讯阅读、交流分享为一体的网络公益平台,不涉及商业盈利目的。如涉及版权问题,请联系我们予以删除。
感谢:重医大附一院检验科及江北区人民医院提供的内容。

相关文章
热门文章

热门问题

